The Prometheus League
Breaking News and Updates
- Abolition Of Work
- Ai
- Alt-right
- Alternative Medicine
- Antifa
- Artificial General Intelligence
- Artificial Intelligence
- Artificial Super Intelligence
- Ascension
- Astronomy
- Atheism
- Atheist
- Atlas Shrugged
- Automation
- Ayn Rand
- Bahamas
- Bankruptcy
- Basic Income Guarantee
- Big Tech
- Bitcoin
- Black Lives Matter
- Blackjack
- Boca Chica Texas
- Brexit
- Caribbean
- Casino
- Casino Affiliate
- Cbd Oil
- Censorship
- Cf
- Chess Engines
- Childfree
- Cloning
- Cloud Computing
- Conscious Evolution
- Corona Virus
- Cosmic Heaven
- Covid-19
- Cryonics
- Cryptocurrency
- Cyberpunk
- Darwinism
- Democrat
- Designer Babies
- DNA
- Donald Trump
- Eczema
- Elon Musk
- Entheogens
- Ethical Egoism
- Eugenic Concepts
- Eugenics
- Euthanasia
- Evolution
- Extropian
- Extropianism
- Extropy
- Fake News
- Federalism
- Federalist
- Fifth Amendment
- Fifth Amendment
- Financial Independence
- First Amendment
- Fiscal Freedom
- Food Supplements
- Fourth Amendment
- Fourth Amendment
- Free Speech
- Freedom
- Freedom of Speech
- Futurism
- Futurist
- Gambling
- Gene Medicine
- Genetic Engineering
- Genome
- Germ Warfare
- Golden Rule
- Government Oppression
- Hedonism
- High Seas
- History
- Hubble Telescope
- Human Genetic Engineering
- Human Genetics
- Human Immortality
- Human Longevity
- Illuminati
- Immortality
- Immortality Medicine
- Intentional Communities
- Jacinda Ardern
- Jitsi
- Jordan Peterson
- Las Vegas
- Liberal
- Libertarian
- Libertarianism
- Liberty
- Life Extension
- Macau
- Marie Byrd Land
- Mars
- Mars Colonization
- Mars Colony
- Memetics
- Micronations
- Mind Uploading
- Minerva Reefs
- Modern Satanism
- Moon Colonization
- Nanotech
- National Vanguard
- NATO
- Neo-eugenics
- Neurohacking
- Neurotechnology
- New Utopia
- New Zealand
- Nihilism
- Nootropics
- NSA
- Oceania
- Offshore
- Olympics
- Online Casino
- Online Gambling
- Pantheism
- Personal Empowerment
- Poker
- Political Correctness
- Politically Incorrect
- Polygamy
- Populism
- Post Human
- Post Humanism
- Posthuman
- Posthumanism
- Private Islands
- Progress
- Proud Boys
- Psoriasis
- Psychedelics
- Putin
- Quantum Computing
- Quantum Physics
- Rationalism
- Republican
- Resource Based Economy
- Robotics
- Rockall
- Ron Paul
- Roulette
- Russia
- Sealand
- Seasteading
- Second Amendment
- Second Amendment
- Seychelles
- Singularitarianism
- Singularity
- Socio-economic Collapse
- Space Exploration
- Space Station
- Space Travel
- Spacex
- Sports Betting
- Sportsbook
- Superintelligence
- Survivalism
- Talmud
- Technology
- Teilhard De Charden
- Terraforming Mars
- The Singularity
- Tms
- Tor Browser
- Trance
- Transhuman
- Transhuman News
- Transhumanism
- Transhumanist
- Transtopian
- Transtopianism
- Ukraine
- Uncategorized
- Vaping
- Victimless Crimes
- Virtual Reality
- Wage Slavery
- War On Drugs
- Waveland
- Ww3
- Yahoo
- Zeitgeist Movement
-
Prometheism
-
Forbidden Fruit

-
The Evolutionary Perspective

Category Archives: Genome
Scientists are on a path to sequencing 1 million human genomes and use big data to unlock genetic secrets – GCN.com
Posted: April 19, 2021 at 7:18 am
Scientists are on a path to sequencing 1 million human genomes and use big data to unlock genetic secrets
The first draft of the human genome waspublished 20 years agoin2001, took nearly three years and costbetween US$500 million and $1 billion. TheHuman Genome Projecthas allowed scientists to read, almost end to end, the 3 billion pairs of DNA bases or letters that biologically define a human being.
That project has allowed a new generation ofresearchers like me, currently a postdoctoral fellow at the National Cancer Institute, to identifynovel targets for cancer treatments, engineermice with human immune systemsand even build awebpage where anyone can navigate the entire human genomewith the same ease with which you use Google Maps.
The first complete genome was generated from a handful of anonymous donors to try to produce a reference genome that represented more than just one single individual. But this fell far short of encompassingthe wide diversity of human populations in the world. No two people are the same and no two genomes are the same, either. If researchers wanted to understand humanity in all its diversity, it would take sequencing thousands or millions of complete genomes. Now, a project like that is underway.
Understanding genetic diversity
The wealth of genetic variation among people is what makes each person unique. But genetic changes also cause many disorders and make some groups of people more susceptible to certain diseases than others.
Around the time of the Human Genome Project, researchers were also sequencing the complete genomes of organisms such asmice,fruit flies,yeastsandsome plants. The huge effort made to generate these first genomes led to a revolution in the technology required to read genomes. Thanks to these advances, instead of taking years and costing hundreds of millions of dollars to sequence a whole human genome, it now takesa few days and costs merely a thousand dollars. Genome sequencing is very different from genotyping services like 23 and Me or Ancestry, which look at only a tiny fraction of locations in a persons genome.
Advances in technology have allowed scientists to sequence the complete genomes of thousands of individuals from around the world. Initiatives such as theGenome Aggregation Consortiaare currently making efforts to collect and organize this scattered data. So far, that group has been able to gather nearly150,000 genomesthat show an incredible amount of human genetic diversity. Within that set, researchers have found more than 241 million differences in peoples genomes,with an average of one variant for every eight base pairs.
Most of these variations are very rare and will have no effect on a person. However, hidden among them are variants with important physiological and medical consequences. For example, certain variants in the BRCA1 gene predispose some groups of woman, like Ashkenazi Jews, toovarian and breast cancer. Other variants in that gene lead someNigerian women to experience higher-than-normal mortalityfrom breast cancer.
The best way researchers can identify these types of population-level variants is throughgenomewide association studiesthat compare the genomes of large groups of people with a control group. But diseases are complicated. An individuals lifestyle, symptoms and time of onset can vary greatly, and the effect of genetics on many diseases is hard to distinguish. The predictive power of current genomic research is too low to tease out many of these effects becausethere isnt enough genomic data.
Understanding the genetics of complex diseases, especially those related to the genetic differences among ethnic groups, is essentially a big data problem. And researchers need more data.
1,000,000 genomes
To address the need for more data, the National Institutes of Health has started a program calledAll of Us. The project aims to collect genetic information, medical records and health habits from surveys and wearables of more than a million people in the U.S. over the course of 10 years. It also has a goal of gathering more data from underrepresented minority groups to facilitate the study of health disparities. TheAll of Us projectopened to public enrollment in 2018, and more than 270,000 people have contributed samples since. The project is continuing to recruit participants from all 50 states. Participating in this effort are many academic laboratories and private companies.
Posted in Genome
Comments Off on Scientists are on a path to sequencing 1 million human genomes and use big data to unlock genetic secrets – GCN.com
What we can learn from sequencing 1 million human genomes with big data – The Next Web
Posted: at 7:18 am
The first draft of the human genome was published 20 years ago in 2001, took nearly three years and cost between US$500 million and $1 billion. The Human Genome Project has allowed scientists to read, almost end to end, the 3 billion pairs of DNA bases or letters that biologically define a human being.
That project has allowed a new generation of researchers like me, currently a postdoctoral fellow at the National Cancer Institute, to identify novel targets for cancer treatments, engineer mice with human immune systems and even build a webpage where anyone can navigate the entire human genome with the same ease with which you use Google Maps.
The first complete genome was generated from a handful of anonymous donors to try to produce a reference genome that represented more than just one single individual. But this fell far short of encompassing the wide diversity of human populations in the world. No two people are the same and no two genomes are the same, either. If researchers wanted to understand humanity in all its diversity, it would take sequencing thousands or millions of complete genomes. Now, a project like that is underway.
The wealth of genetic variation among people is what makes each person unique. But genetic changes also cause many disorders and make some groups of people more susceptible to certain diseases than others.
Around the time of the Human Genome Project, researchers were also sequencing the complete genomes of organisms such as mice, fruit flies, yeasts and some plants. The huge effort made to generate these first genomes led to a revolution in the technology required to read genomes. Thanks to these advances, instead of taking years and costing hundreds of millions of dollars to sequence a whole human genome, it now takes a few days and costs merely a thousand dollars. Genome sequencing is very different from genotyping services like 23 and Me or Ancestry, which look at only a tiny fraction of locations in a persons genome.
Advances in technology have allowed scientists to sequence the complete genomes of thousands of individuals from around the world. Initiatives such as the Genome Aggregation Consortia are currently making efforts to collect and organize this scattered data. So far, that group has been able to gather nearly 150,000 genomes that show an incredible amount of human genetic diversity. Within that set, researchers have found more than 241 million differences in peoples genomes, with an average of one variant for every eight base pairs.
Most of these variations are very rare and will have no effect on a person. However, hidden among them are variants with important physiological and medical consequences. For example, certain variants in the BRCA1 gene predispose some groups of woman, like Ashkenazi Jews, to ovarian and breast cancer. Other variants in that gene lead some Nigerian women to experience higher-than-normal mortality from breast cancer.
The best way researchers can identify these types of population-level variants is through genomewide association studies that compare the genomes of large groups of people with a control group. But diseases are complicated. An individuals lifestyle, symptoms and time of onset can vary greatly, and the effect of genetics on many diseases is hard to distinguish. The predictive power of current genomic research is too low to tease out many of these effects because there isnt enough genomic data.
Understanding the genetics of complex diseases, especially those related to the genetic differences among ethnic groups, is essentially a big data problem. And researchers need more data.
The link between genetics and disease is nuanced, but the more genomes you can study, the easier it is to find those links. Image via brian0918/Wikimedia Commons
To address the need for more data, the National Institutes of Health has started a program called All of Us. The project aims to collect genetic information, medical records and health habits from surveys and wearables of more than a million people in the U.S. over the course of 10 years. It also has a goal of gathering more data from underrepresented minority groups to facilitate the study of health disparities. The All of Us project opened to public enrollment in 2018, and more than 270,000 people have contributed samples since. The project is continuing to recruit participants from all 50 states. Participating in this effort are many academic laboratories and private companies.
This effort could benefit scientists from a wide range of fields. For instance, a neuroscientist could look for genetic variations associated with depression while taking into account exercise levels. An oncologist could search for variants that correlate with reduced risk of skin cancer while exploring the influence of ethnic background.
A million genomes and the accompanying health and lifestyle information will provide an extraordinary wealth of data that should allow researchers to discover the effects of genetic variation on diseases, not only for individuals, but also within different groups of people.
[Understand new developments in science, health and technology, each week.Subscribe to The Conversations science newsletter.]
Another benefit of this project is that it will allow scientists to learn about parts of the human genome that are currently very hard to study. Most genetic research has been on the parts of the genome that encode for proteins. However, these represent only 1.5% of the human genome.
My research focuses on RNA a molecule that turns the messages encoded in a persons DNA into proteins. However, RNAs that come from the 98.5% of the human genome that doesnt make proteins have a myriad of functions by themselves. Some of these noncoding RNAs are involved in processes such as how cancer spreads, embryonic development or controlling the X chromosome in females. In particular, I study how genetic variations can influence the intricate folding that allows noncoding RNAs to do their jobs. Since the All of Us project includes all coding and noncoding parts of the genome, it is going to be by far the largest dataset relevant to my work and will hopefully shed light on these mysterious RNAs.
The first human genome sparked 20 years of incredible scientific progress. I think it is almost certain that a huge dataset of genomic variations will unlock clues about complex diseases. Thanks to large-scale population studies and big-data projects such as All of Us, researchers are paving the way to answering, in the next decade, how our individual genetics shape our health.
This article byXavier Bofill De Ros, Research Fellow in RNA biology, National Institutes of Health,is republished from The Conversation under a Creative Commons license. Read the original article.
Read more:
What we can learn from sequencing 1 million human genomes with big data - The Next Web
Posted in Genome
Comments Off on What we can learn from sequencing 1 million human genomes with big data – The Next Web
The Nike Air Max Genome Receives A Sharp And Simple Black And White – Sneaker News
Posted: at 7:18 am
Previewed ahead of the Swooshs fictitious March 26th holiday, the Nike Air Max Genome is set to take over the casual footwear space as it delivers comfortable solutions clad in versatile colorways.
Unlike some of the inaugural options, a forthcoming pair indulges in a simple, but sharp Black/White color palette. The mix of synthetic, textile and fabric across the upper harkens back to the early 2000s, while simultaneously fitting into the current sneaker landscape. Branding throughout the shoe delivers stark White contrast, which is highlighted at the midsole. Full-length Air Max cushioning, however, opts into a semi-opaque arrangement. Lastly, tread reverts to an understated Black color.
An official Nike.com release date is unknown, but this Air Max Genome is likely to quietly launch soon. In the meantime, enjoy images of the pair here below.
For more styles from NIKE, Inc., check out the new Jordan Delta 2.
Where to Buy
Make sure to follow @kicksfinder for live tweets during the release date.
Mens: $170Style Code: CW1648-003
Images: Nike
Read more here:
The Nike Air Max Genome Receives A Sharp And Simple Black And White - Sneaker News
Posted in Genome
Comments Off on The Nike Air Max Genome Receives A Sharp And Simple Black And White – Sneaker News
Second Genome to Present at the Jefferies Microbiome-Based Therapeutics Summit – PRNewswire
Posted: at 7:18 am
BRISBANE, Calif., April 15, 2021 /PRNewswire/ --Second Genome, a tech-enabled biotechnology company that extracts microbial genetic insights to make transformational precision therapies and biomarkers, today announced that Karim Dabbagh, Ph.D., President and Chief Executive Officer, will present and participate in a fireside chat at the virtual Jefferies Microbiome-Based Therapeutics Summit on April 22, 2021.
The prerecorded presentation and fireside chat will be available on Thursday, April 22, 2021, at 8:00 a.m. ET and can be accessed by visiting the "News" section of the Company's website at http://www.secondgenome.com and selecting the Events tab on the News page. A replay of the webcast will be archived there following the presentation date.
About Second Genome
Second Genome is a tech-enabled biotechnology company that extracts microbial genetic insights to make transformational precision therapies and biomarkers through clinical development and commercialization. We built a proprietary microbiome-based drug discovery and development platform with machine-learning analytics, customized protein engineering techniques, phage library screening, mass spec analysis and CRISPR, that we couple with traditional drug development approaches to progress the development of therapies and diagnostics for wide-ranging diseases. Second Genome is advancing deep drug discovery and biomarker pipelines with precision therapeutics and biomarker programs in inflammatory bowel disease (IBD) and cancer, with the lead program SG-2-0776 in IBD expected to enter clinical development in 2022. We also collaborate with industry, academic and governmental partners to leverage our microbiome platform and data science. We hold a strategic collaboration with Gilead Sciences, Inc., utilizing our proprietary platform and comprehensive data sets to identify novel biomarkers associated with clinical response to Gilead's investigational medicines. For more information, please visit http://www.secondgenome.com.
Investor Contact:Argot Partners212-600-1902[emailprotected]
Media Contact:Argot Partners212-600-1902[emailprotected]
SOURCE Second Genome
secondgenome.com
View original post here:
Second Genome to Present at the Jefferies Microbiome-Based Therapeutics Summit - PRNewswire
Posted in Genome
Comments Off on Second Genome to Present at the Jefferies Microbiome-Based Therapeutics Summit – PRNewswire
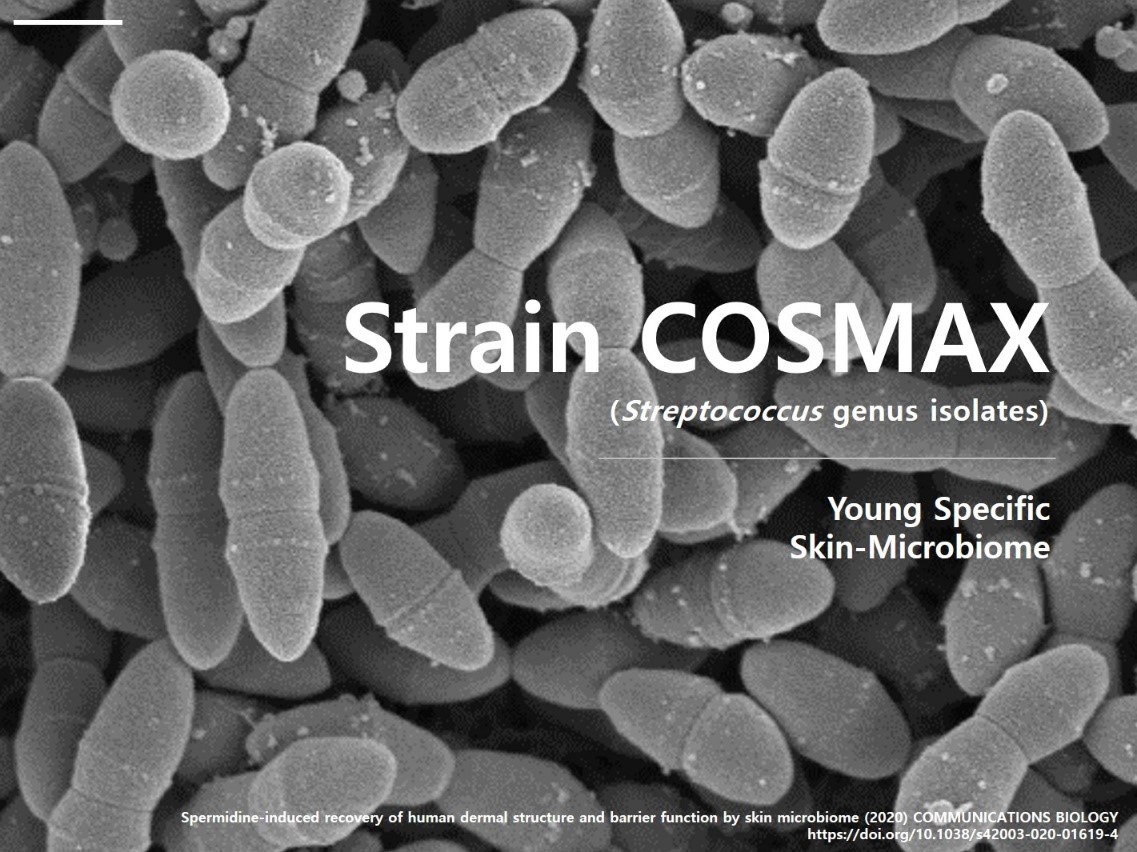
Hankyung.com’s Introduces: COSMAX’s Skin Microbiome Research Becomes First of Its Kind to Be Published in International Scientific Journal -…
Posted: at 7:18 am
SEOUL, South Korea, April 19, 2021 /PRNewswire/ -- Global cosmetics research, development, and ODM company, COSMAX (CEO Lee Byung-man), and the Gwangju Institute of Science and Technology (GIST) have become the first in the world to reveal the correlation between the skin microbiome and skin aging.
COSMAX announced that its thesis titled, "Spermidine-induced Recovery of Dermal Structure and Barrier Function by Skin Microbiome," has been published by Nature Communications Biology.
In 2015, COSMAX identified that a person's skin flora directly regulates the skin, and the company began to carry out research related to anti-aging. After discovering that the number of microorganisms that live on the skin decreases with age, they hypothesized that it held an important key to anti-aging and started performing genetic analysis.
They named a newly-discovered microorganism "Strain-COXMAX," and together with GIST, they conducted a whole genome analysis that can trace the role of entire genomes to uncover the microorganism's anti-aging functions.
The results of the analysis revealed that the microorganism affects the aging process by regulating various metabolic functions of the skin. It was also discovered that spermidine, which is created during the metabolic process, directly impacts skin anti-aging. Furthermore, spermidine showed efficacy in improving skin moisture, elasticity, and anti-aging by activating collagenisis and lipid secretion.
"Discovering the correlation mechanism between the skin microbiome and aging is a feat that was achieved through six years of hard work," said R&I center director Park Myeong-sam. "The technology super gap will be used in the next generation of anti-aging cosmetics and biomaterials in the global market."
COXMAX is expected to expand the application of the skin microbiome to various products. Such products include:
"The publishing of skin microbiome technology in a global scientific journal creates an opportunity for the R&D status of K-beauty to be promulgated," said COSMAX CEO Lee Byung-man. "It will take center stage in the global health and beauty market by using innovative materials to develop products that don't yet exist in the world."
In 2019, COSMAX launched the world's first anti-aging cosmetics that utilize Strain-COXMAX, a culture medium with beneficial skin bacteria. By securing more than 20 beneficial microorganisms, the company has become a leader in the skin microbiome market.
https://img.hankyung.com/pdsdata/pr.hankyung.com/uploads/2021/04/20210413COSMAX.jpg< i>taken atx100000 magnification by SEM (scanning electronic microscope)>
Microbiome, which is a portmanteau of "microbe" and "biome," describes the microorganisms that inhabit the human body, as well as their genomes. The microbiome is so vast that it makes up 1 to 3 percent of the body's mass, and it has over 100 times more genes than our own genome. Called the "second genome," the microbiome could be seen as an ecosystem that holds a great deal of information about the body.
As it regulates immune functions and forms various kinds of metabolites, the microbiome is known to influence obesity, diabetes, atopic dermatitis, cancer, and autoimmune diseases. Thus, research on the exact role of the microbiome in disease and aging has greatly increased, and related markets have been experiencing rapid growth.
Since its establishment in 2015, Genome & Company has become a global leader in microbiome immuno-oncology. It focuses on developing the next waves of innovative therapeutics in immune-oncology through diverse modalities of microbiome and novel target immune checkpoint inhibitors.
SOURCE Hankyung.com
Read this article:
Hankyung.com's Introduces: COSMAX's Skin Microbiome Research Becomes First of Its Kind to Be Published in International Scientific Journal -...
Posted in Genome
Comments Off on Hankyung.com’s Introduces: COSMAX’s Skin Microbiome Research Becomes First of Its Kind to Be Published in International Scientific Journal -…
Whole-Genome Sequencing as an Alternative to Cytogenetics in AML, MDS – Oncology Learning Network
Posted: at 7:17 am
Whole-genomesequencing (WGS) could be an alternative to conventional cytogenetic analysis in patients with acute myeloid leukemia (AML) or myelodysplastic syndromes (MDS), according to a study published in the New England Journal of Medicine (N Engl J Med. 2021;384[10]:924-935).
Genomic analysis is essential for risk stratification in patients with AML or MDS, said Eric J. Duncavage, MD, Department of Pathology and Immunology, McDonnell Genome Institute, and the Divisions of Oncology and Biostatistics, Department of Medicine, Washington University School of Medicine, St. Louis, and colleagues.
DrDuncavage and colleaguesobtained the genomic profiles of 263 patients with myeloid cancers, including 235 patients who had undergone cytogenetic analysis. Using WGS, they detected all 40 recurrent translocations and 91 copy-number alterations that the cytogenetic analysis identified. In 40 patients (17%, n = 235), new clinically reportable genomic events were found.
Prospective sequencing was performed on samples taken from 117 consecutive patients over a median of 5 days and provided new genetic information in 29 patients (24.8%), which in turn, changed the risk category for 19 patients (16.2%).
The researchers concluded thatWGS provided rapid and accurate genomic profiling in patients with AML or MDS.Emily Bader
Follow this link:
Whole-Genome Sequencing as an Alternative to Cytogenetics in AML, MDS - Oncology Learning Network
Posted in Genome
Comments Off on Whole-Genome Sequencing as an Alternative to Cytogenetics in AML, MDS – Oncology Learning Network
Bionano Genomics Announces Record Number of Presentations on Optical Genome Mapping and Structural Variation at the 2021 Annual Clinical Genetics…
Posted: April 13, 2021 at 6:42 am
Sixteen talks and posters, fourteen of which are from US institutions, to be presented across all four of Bionanos main target growth markets: prenatal, postnatal/constitutional genetics, blood cancers and solid tumor analysis
SAN DIEGO, April 12, 2021 (GLOBE NEWSWIRE) -- Bionano Genomics, Inc. (Nasdaq: BNGO) today announced it largest presence to date at the 2021 Annual Clinical Genetics Meeting of the American College of Medical Genetics and Genomics (ACMG), which is being held in a virtual format from April 13-16, 2021. The meeting features a total of sixteen presentations by Saphyr customers and Bionano scientists, almost three times the number presented last year and nearly all based on work done in the United States.
ACMG this year has the greatest number of presentations featuring Bionano data to date, commented Erik Holmlin, PhD, CEO of Bionano Genomics. We believe this multitude of presentations shows how significant Bionanos presence and that of optical genome mapping (OGM) has become in the medical genetics community. The progress we are seeing here from the US market, where almost all the data being presented at ACMG comes from, is particularly encouraging since the US has a higher barrier to adoption that could be addressed with data outlining the value proposition of our products and technology. We are thrilled to share this important clinical data in all four main target segments (prenatal, postnatal/constitutional genetics, blood cancers and solid tumor analysis) at the meeting.
Blood Cancers Oral and Poster Presentations:
Platform Presentation, Thursday, April 15, 2021, 5:30 pm. OP340 Optical Genome Mapping for Assessment of Genomic Aberrations in Acute Myeloid Leukemia: A Multicenter Evaluation
eP085 Efficient workflow for detection of clinically relevant abnormalities in leukemias according to NCCN guidelines
eP068 Optical Genome Mapping Detects Rare Genetic Drivers in Pediatric B-Lymphoblastic Leukemia
Prenatal Oral and Poster Presentations:
Story continues
Product Theater (available on demand): Next-Generation Cytogenomic Characterization of Prenatal Cases by Optical Genome Mapping
eP501 Generation of a Prenatal Workflow for Identification of Structural Variation by Optical Genome Mapping (OGM)
eP502 Next-Generation Cytogenomic Characterization of Prenatal Cases by Optical Genome Mapping
Post-Natal Oral and Poster Presentations:
Platform Presentation, Wednesday, 4/14, 4:15 pm, part of Scientific Concurrent Session Hot Topics: Pushing the Boundaries of Genome Sequencing Clinically Relevant Genes Hiding In Plain Sight And How Long Range Technologies Resolve Them
eP436 Identification of Structural Variation in Constitutional Disorders by Optical Genome Mapping
eP406 Optical Genome Mapping Enables Constitutional Chromosomal Aberration Detection: Proof-of-Principle Study with 85 Samples
eP294 High Throughput Analysis of Disease Repeat Expansions and Contractions by Optical Mapping
eP447 Improving and Accelerating Clinical Molecular Diagnosis of Severe Hemophilia A with Optical Genome Mapping Technology
eP370 Fascioscapulohumeral Muscular Dystrophy Genetic Testing by Optic Mapping
Solid Tumors Poster Presentations:
eP083 Clinical Utility of Optical Genome Mapping (OGM) in Cytogenetic Analysis of Brain Tumors
eP388 Optical Genomic Mapping Reveals Balanced and Unbalanced Cytogenetic Findings Associated with Tumor-forming Potential in a Prostate Cancer Cell line (M2205)
Other Bionano Poster Presentations:
eP407 NeuroSCORE: A Genome-wide OMICs Based Model to Identify Disease Associated Genes of the Central Nervous System
eP365 PRKX/PRKY-Mediated Xp;Yp Translocations: A Significant Contributor to SRY-Positive 46,XX TDSD and Potential Risk of Recurrence in Common Yp Inversion Carriers
More details can be found at https://www.acmgmeeting.net/acmg2021/Public/mainhall.aspx
About the ACMG Meeting
The ACMG Meeting is the genetics meeting most focused specifically on the practical applications of genetic discoveries to clinical medicine. Topics range from common conditions to rare diseases. The ACMG Annual Meeting attracts medical and scientific leaders from around the world who are working to apply research in genetics and the human genome to the diagnosis, management, treatment and prevention of genetic conditions and rare and common diseases in patients in the clinical setting.
About Bionano Genomics
Bionano is a genome analysis company providing tools and services based on its Saphyr system to scientists and clinicians conducting genetic research and patient testing, and providing diagnostic testing for those with autism spectrum disorder (ASD) and other neurodevelopmental disabilities through its Lineagen business. Bionanos Saphyr system is a research use only platform for ultra-sensitive and ultra-specific structural variation detection that enables researchers and clinicians to accelerate the search for new diagnostics and therapeutic targets and to streamline the study of changes in chromosomes, which is known as cytogenetics. The Saphyr system is comprised of an instrument, chip consumables, reagents and a suite of data analysis tools. Bionano provides genome analysis services to provide access to data generated by the Saphyr system for researchers who prefer not to adopt the Saphyr system in their labs. Lineagen has been providing genetic testing services to families and their healthcare providers for over nine years and has performed over 65,000 tests for those with neurodevelopmental concerns. For more information, visit http://www.bionanogenomics.com or http://www.lineagen.com.
Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as may, will, expect, plan, anticipate, estimate, intend and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) convey uncertainty of future events or outcomes and are intended to identify these forward-looking statements. Forward-looking statements include statements regarding our intentions, beliefs, projections, outlook, analyses or current expectations concerning, among other things: the timing and content of the presentations identified in this press release; our presence in the medical genetics community; our ability to address barriers to adoption in the United States; and the execution of Bionanos strategy, including with respect to our target growth markets. Each of these forward-looking statements involves risks and uncertainties. Actual results or developments may differ materially from those projected or implied in these forward-looking statements. Factors that may cause such a difference include the risks and uncertainties associated with: Indalo Bios ability to successfully develop assays on the Saphyr system and/or make its technology widely available in Africa; the impact of the COVID-19 pandemic on our business and the global economy; general market conditions; changes in the competitive landscape and the introduction of competitive products; changes in our strategic and commercial plans; our ability to obtain sufficient financing to fund our strategic plans and commercialization efforts; the ability of medical and research institutions to obtain funding to support adoption or continued use of our technologies; the loss of key members of management and our commercial team; and the risks and uncertainties associated with our business and financial condition in general, including the risks and uncertainties described in our filings with the Securities and Exchange Commission, including, without limitation, our Annual Report on Form 10-K for the year ended December 31, 2020 and in other filings subsequently made by us with the Securities and Exchange Commission. All forward-looking statements contained in this press release speak only as of the date on which they were made and are based on management's assumptions and estimates as of such date. We do not undertake any obligation to publicly update any forward-looking statements, whether as a result of the receipt of new information, the occurrence of future events or otherwise.
CONTACTSCompany Contact:Erik Holmlin, CEOBionano Genomics, Inc.+1 (858) 888-7610eholmlin@bionanogenomics.com
Investor Relations and Media Contact:Amy ConradJuniper Point+1 (858) 366-3243amy@juniper-point.com
Continue reading here:
Bionano Genomics Announces Record Number of Presentations on Optical Genome Mapping and Structural Variation at the 2021 Annual Clinical Genetics...
Posted in Genome
Comments Off on Bionano Genomics Announces Record Number of Presentations on Optical Genome Mapping and Structural Variation at the 2021 Annual Clinical Genetics…
Ark Invest analyst breaks down Adaptive Biotechnologies and Invitae – CNBC
Posted: at 6:42 am
Chad Robins, CEO of Adaptive Biotechnologies.
Anjali Sundaram | CNBC
Ark Invest analyst Simon Barnett on Monday explained the firm's approach to what it calls the genomic revolution, breaking down two of its favorite holdings: Adaptive Biotechnologies and Invitae.
In an interview on CNBC's "Closing Bell," Barnett said the Cathie Wood-led Ark Invest sees major potential for investors in the branch of molecular biology known as genomics, calling it "one of the most transformative investment opportunities of the century."
Ark's family of funds includes the Genomic Revolution ETF (ARKG), which seeks to offer investors exposure to areas such as DNA sequencing technology and molecular diagnostics.
ARKG is down about 7% year to date.
However, in the past 12 months, the ETF has risen roughly 160%. The firm's flagship fund is theArk Innovation ETF(ARKK), which has fallen about 1.6% so far in 2021. ARKK also is up nearly 160% in the past 12 months.
View original post here:
Ark Invest analyst breaks down Adaptive Biotechnologies and Invitae - CNBC
Posted in Genome
Comments Off on Ark Invest analyst breaks down Adaptive Biotechnologies and Invitae – CNBC
Global Whole Genome and Exome Sequencing Markets Report 2021-2025 Including Updated Whole Genome Sequence of Sars-Cov-2 – ResearchAndMarkets.com -…
Posted: at 6:42 am
TipRanks
The investing game is rarely plain sailing. While no doubt investors would like the choices that make up their portfolio to always go up, the reality is more complicated. There are periods when even shares of the worlds most successful companies have been on a downward trajectory for one reason or another. While its no fun watching a stock you own drift to the bottom, any savvy investor knows that if the companys fundamentals are sound to begin with, the pullback is often a gift in disguise. This is where the chance for strong returns really comes into play. Buy the Dip is not a clich without reason. With this in mind, we scoured the TipRanks database and picked out 3 names which have been heading south recently, specifically ones pinpointed by those in the know as representing a buying opportunity. Whats more, all 3 are rated Strong Buys by the analyst consensus and projected to rake in at least 70% of gains over the next 12 months. Here are the details. Flexion Therapeutics (FLXN) Lets first take a look at Flexion, a pharma company specializing in the development and commercialization of therapies for the treatment of musculoskeletal pain. The company has two drugs currently in early-stage clinical trials but one which has already been approved by the FDA; Zilretta is an extended-release corticosteroid for the management of osteoarthritis knee pain. The drug was granted regulatory approval in 2017, and Flexion owns the exclusive worldwide rights. FLXN stock has found 2021 hard going and is down by 30% year-to-date. However, the recent weakness, says Northland analyst Carl Byrnes has created a unique buying opportunity. Like many biopharmas, Flexions marketing efforts took a hit during the height of the pandemic last year, as shutdowns and restrictions impacted its operations. However, Byrnes anticipates Zilretta to exhibit stellar growth in 2021 and beyond. We remain highly confident that the demand for ZILRETTA will continue to strengthen, bolstered by product awareness and positive clinical experiences of both patients and HCP, augmented by improvements in HCP interactions and deferral of total knee arthroplasty (TKA) surgical procedures, the analyst said. Byrnes expects Zilrettas 2021 sales to surge by 45% year-over-year to $125 million, and then increase by a further 50% to $187.5 million the following year. That revenue growth will go hand in hand with massive share appreciation; Byrnes price target is $35, suggesting upside of ~339% over the next 12 months. Needless to say Byrnes rating is an Outperform (i.e. Buy). (To watch Byrnes track record, click here) Barring one lone Hold, all of Byrnes colleagues agree. With 9 Buys, FLXN stock boasts a Strong Buy consensus rating. While not as optimistic as Byrnes objective, the $20.22 average price target is still set to yield returns of an impressive 153% within the 12-month time frame. (See FLXN stock analysis on TipRanks) Protara Therapeutics (TARA) Staying in the pharma industry, next up we have Protara. Unlike Flexion, the cancer and rare disease-focused biotech has no therapies approved yet. However, the picture should soon become clear regarding the timing of a BLA (biologics license application) for TARA-002, the companys investigational cell therapy for a rare pediatric indication - lymphatic malformations (LM). TARA-002 is based on the immunopotentiator OK-432, currently approved as Picibanil in Japan and Taiwan for the treatment of multiple cancer indications as well as LM. Currently, Protara is seeking to get the FDAs acceptance that TARA-002 is comparable to OK-432. If everything goes according to plan, the company anticipates potential BLA filing in H2:2021 and potential approval in H1:2022. Protara shares have tumbled 40% year-to-date. That said, Guggenheim analyst Etzer Darout believes the stock is significantly undervalued. We estimate risk-adjusted peak sales of ~$170M (75% PoS) in the US alone (biologics exclusivity to 2034-2035), the 5-star analyst said. The company has outlined a no additional study scenario that estimates a US launch in 2022 and an additional registration study scenario that estimates a 2023 launch and we see current levels as a buying opportunity ahead of regulatory clarity on LM. Furthermore, Tara is expected to submit an IND (investigational new drug) for a Phase 1 trial for TARA-002 in 2H21 for the treatment of non-muscle invasive bladder cancer (NMIBC). Darout notes 80% (~65K) of all newly diagnosed bladder cancer patients suffer from this specific condition including ~45% that are high grade with high unmet need. The company also owns IV Choline, a Phase 3-ready asset, for which the FDA has already granted both Orphan Drug Designation and Fast Track Designation for IFALD (intestinal failure-associated liver disease). Based on all of the above, Darout rates TARA a Buy and has a $48 price target for the shares. The implication for investors? Upside of a strong 225%. (To watch Darouts track record, click here) Overall, with 3 recent Buy ratings under its belt, TARA gets a Strong Buy from the analyst consensus view. The stock is backed by an optimistic average price target, too; at $43.67, the shares are anticipated to appreciate by ~198% in the year ahead. (See TARA stock analysis on TipRanks) Green Thumb Industries (GTBIF) Last but not least is Green Thumb, a leading US cannabis MSO (multi state operator). This Chicago-based company is one of the stalwarts of the rising cannabis sector, boasting the second highest market-cap in the industry and exhibiting impressive growth over the last year. In 2020, revenue increased by 157% from 2019, to reach $556.6 million. That said, despite delivering another excellent quarterly statement in March, and being well-positioned to capitalize on additional states legalizing cannabis, the stock has pulled back recently after the company was hit by a damning Chicago Tribune article. According to Chicago Tribune, the company is being investigated by the fed over "pay to play" payments regarding the procurement of cannabis licenses in Illinois. Countering the claims, GTBIF management said the allegations are unfounded and that there is no factual evidence to support them. Furthermore, the company pointed out it has not even been contacted by the authorities regarding the matter. Who to believe, then? Its an easy choice, according to Roth Capitals Scott Fortune. We believe these tenuous claims create an opportunity to own the best-in-class operator currently off 25% from recent highs, the 5-atar analyst opined. In our view, the GTI business and track record of execution is not at risk in terms of the seemingly baseless accusations. We will continue to monitor any new additional incremental evidence potentially surfacing but believe the allegations are unfounded. We believe the upside opportunity remains compelling at these levels. Going by Fortunes $45 price target, shares will be changing hands for a 70% premium a year from now. Fortunes rating remains a Buy. (To watch Fortunes track record, click here) The negative news has done little to dampen enthusiasm around this stock on Wall Street. The analyst consensus rates GTBIF a Strong Buy, based on a unanimous 12 Buys. The average price target, at $47.71, suggests an upside of 79% over the next 12 months. (See GTBIF stock analysis on TipRanks) To find good ideas for stocks trading at attractive valuations, visit TipRanks Best Stocks to Buy, a newly launched tool that unites all of TipRanks equity insights. Disclaimer: The opinions expressed in this article are solely those of the featured analysts. The content is intended to be used for informational purposes only. It is very important to do your own analysis before making any investment.
Follow this link:
Global Whole Genome and Exome Sequencing Markets Report 2021-2025 Including Updated Whole Genome Sequence of Sars-Cov-2 - ResearchAndMarkets.com -...
Posted in Genome
Comments Off on Global Whole Genome and Exome Sequencing Markets Report 2021-2025 Including Updated Whole Genome Sequence of Sars-Cov-2 – ResearchAndMarkets.com -…
Genomic testing services in pathology & immunology department to be expanded Washington University School of Medicine in St. Louis – Washington…
Posted: at 6:42 am
Visit the News Hub
Changes aimed at improving diagnosis of cancer, inherited diseases
The Department of Pathology & Immunology at Washington University School of Medicine in St. Louis is reorganizing and expanding its genomic medicine testing services to take advantage of advances in genetic and genomic sciences and improve clinical care.
Genetic and genomic testing is driving advances in precision medicine. Such testing provides the data that, when combined with information about disease status and environmental factors, enable doctors to move away from one-size-fits-all treatment plans to personalized therapies tailored to individual needs.
The Department of Pathology & Immunology at Washington University School of Medicine in St. Louis is reorganizing and expanding its genomic medicine testing services to take advantage of advances in genetic and genomic sciences and improve clinical care. Several new tests for cancer and inherited diseases are in the works. The department also is redesigning its website to make it easier for physicians to quickly identify the most appropriate tests for their patients.
As part of the reorganization, the department is establishing two new sections: a molecular oncology section, led by Eric Duncavage, MD, and an inherited diseases section, headed by Jonathan Heusel, MD, PhD.
The School of Medicine has been at the forefront of genetics research for many years, including leadership in whole genome sequencing, saidRichard Cote, MD, the Edward Mallinckrodt Professor and head of the Department of Pathology & Immunology. We have long had a major stake in translating these discoveries into clinically actionable tests to better define prognosis and treatment for a wide variety of diseases. We are delighted that Jon Heusel and Eric Duncavage, widely recognized for their research, are leading the effort to enhance the departments genetic and genomic testing services.
A wide variety of different mutations can give rise to cancer, and the specific mutations carried by a particular tumor affect its susceptibility to a particular treatment. These mutations also can be used to track a tumor during therapy to determine how it is responding to therapy. Clinical cancer genomics aims to use information on tumor mutations to help identify the therapies most likely to benefit the patient and avoid those least likely to help.
Duncavage
Duncavage and his Washington University colleagues recently developed a diagnostic test, ChromoSeq, for blood cancers, based on sequencing the whole genome. This comprehensive analysis has greater sensitivity than traditional testing, providing additional information that could help clinicians assess each patients risk of severe disease and choose the best treatment plan. The test was evaluated in a clinical trial of patients treated at Siteman Cancer Center, based at Barnes-Jewish Hospital and Washington University School of Medicine. Results of that trial were published in March in The New England Journal of Medicine. ChromoSeq whole genome sequencing for blood cancers will be available as a clinical test through the Department of Pathology & Immunology.
Until recently, we could only analyze a limited set of genes or chromosomes because whole genome sequencing was too expensive for routine clinical use, Duncavage said. But recent advances in sequencing technologies and data analysis techniques have driven down the cost of whole genome sequencing in both time and resources. We showed that the technology could be used for blood cancers, and we are working on applying it to other kinds of cancers.
In addition, Heusel and Duncavage are spearheading an effort to develop tests to identify people with an inherited predisposition to cancer. Most famously, variations in the BRCA1 and BRCA2 genes increase the risk of breast and ovarian cancer, but dozens of other genes have been linked to a range of cancers. In the interest of efficiency, the tests under development are based on sequencing only the 2% of the genome that codes for proteins. This tiny fraction of the genome known as the exome is where nearly all mutations associated with disease risk are found.
Heusel
As chief of the inherited diseases section, Heusel will oversee the development and operation of tests for rare genetic diseases. Some of the people most in need of whole genome testing are babies born with mysterious ailments that appear to have genetic causes. Sequencing an affected newborns entire genome and often the genomes of both parents, too can be the fastest way to find an explanation for a babys condition.
Heusel and colleagues also are working on an improved test for disorders of somatic mosaicism, a group of conditions characterized by mutations in some cells but not others. Patients can have a wide range of symptoms, including overgrowth of one part of the body, such as the hand; skin spots or rashes; and abnormal tangles of blood vessels. The symptoms depend not only on what mutation has occurred but in which cells it has occurred. The researchers are building a faster, cheaper test that will allow more genes and more conditions to be screened.
Genetic testing particularly what we call next-generation sequencing, which involves sequencing dozens or hundreds of genes or whole exomes or whole genomes its transforming medicine, transforming the way we understand the basis of disease, how to diagnose it and how to treat it, Heusel said. What were really trying to do in the department now is make it easier for clinicians to take advantage of all the new genetic and genomic diagnostic tools that are becoming available.
Washington University School of Medicines 1,500 faculty physicians also are the medical staff of Barnes-Jewish and St. Louis Childrens hospitals. The School of Medicine is a leader in medical research, teaching and patient care, consistently ranking among the top medical schools in the nation by U.S. News & World Report. Through its affiliations with Barnes-Jewish and St. Louis Childrens hospitals, the School of Medicine is linked to BJC HealthCare.
See the article here:
Genomic testing services in pathology & immunology department to be expanded Washington University School of Medicine in St. Louis - Washington...
Posted in Genome
Comments Off on Genomic testing services in pathology & immunology department to be expanded Washington University School of Medicine in St. Louis – Washington…








{kind=link}